
Spektrum genodermatózy je široké. Některé z nich, jako je psoriáza, mohou být relativně mírné a mohou být léčeny lokálními léčbami. Jiné, jako je ichtyóza typu harlekýn, mohou být smrtelné. Níže je pět dědičných kožních problémů, které postihují většinu epidermis.
Darieu-White nemoc
Darieu-Whiteova choroba, klinicky známá jako folikulární keratóza, byla poprvé identifikována na konci 19. století dermatology Ferdinandem-Jeanem Darierem a Jamesem Clarkem Whitem. White identifikoval poruchu jako dědičnou kožní poruchu, když se k němu matka i dcera přišly léčit.
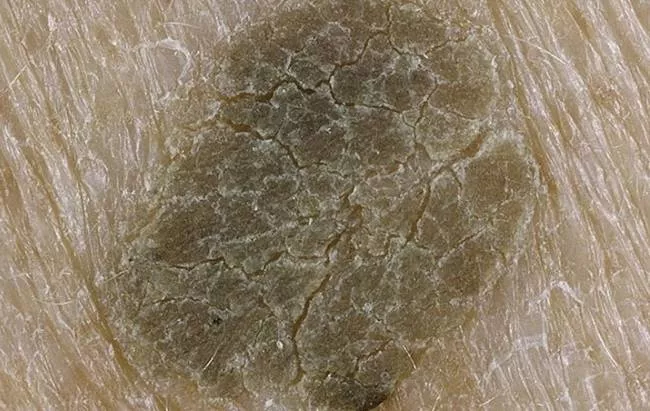
Darieu-Whiteova choroba je charakterizována abnormálním ztvrdnutím kožních buněk na vnější vrstvě kůže, procesem zvaným zrohovatění. Jedná se o stejný proces, který mění zdravé kožní buňky na nehty, ale u lidí s Darier-Whiteovou chorobou se to děje jinde v kůži. Vědci zjistili, že kožní buňky pacientů postrádají sloučeninu, která váže buňky dohromady (tzv. desmozomy), což může přispět k větší zrohovatění.
Nejnápadnějším příznakem jsou malé, mastné, žlutohnědé papuly podobné pupínkům, které se hromadně tvoří v oblastech kolem mazových žláz.
Výzkumníci našli souvislost mezi mutacemi v genu ATP2A2 a Darier-Whiteovou nemocí, ale také nenašli společný vzorec mutací, který by nemoc definoval. Dokonce i lidé se stejnými mutacemi ATP2A2 mohou vykazovat různé příznaky.
Epidermolysis bullosa
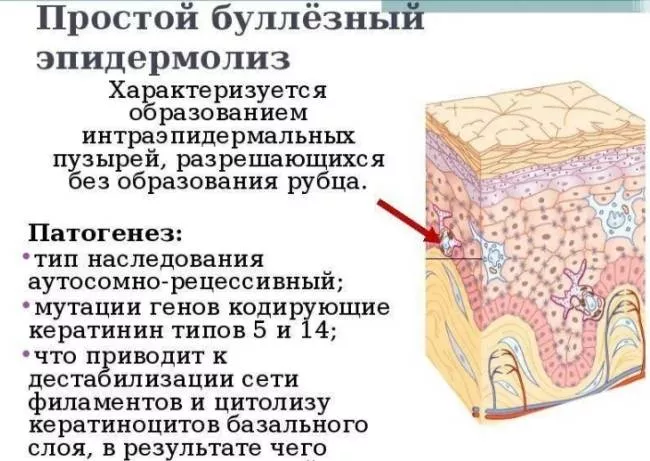
Epidermolysis bullosa (EB) je vysilující dědičná kožní porucha, která má za následek tvorbu puchýřů s mírnými změnami tlaku nebo teploty. Lidé obvykle zaznamenají puchýřkovou reakci, když se vnější a vnitřní vrstva kůže oddělí. Volná oblast je naplněna tekutinou, která působí jako polštář, zatímco se poškozená kůže pod ní hojí.
Pacienti s bulózní epidermolysis mají extrémně zvýšenou reakci na tvorbu puchýřů. Pouhá chůze, plazení, pobyt na rukou a/nebo i mírné změny pokojové teploty mohou způsobit, že se po celé kůži vytvoří bolestivé puchýře. Frekvence tvorby puchýřů zvyšuje pravděpodobnost, že se pacient nakazí infekcí, což vede k dalším zdravotním rizikům, jako je amputace.
Nejméně 10 různých genů bylo spojeno v kombinaci, aby způsobily EB. Ve většině případů je nemoc zděděna od rodičů, i když vzácně může být také výsledkem náhodné mutace.
Nejčastěji se po rodičích dědí mutace v genech odpovědných za expresi keratinových proteinů, které zajišťují strukturu a pevnost vazby mezi vrstvami kůže.
Lamelární ichtyóza
Dědičná choroba lamelární ichtyóza má druhou polovinu svého názvu z latinského slova pro rybu, ichthys. Tento termín je vhodný, protože pacientům s lamelární ichtyózou se v důsledku jejich onemocnění tvoří tlusté šupiny po celém těle.

U zdravých lidí staré buňky chrání mladé buňky před vypadnutím a nahrazením. Pod stratum corneum (vnější vrstva) se kožní buňky známé jako keratinocyty dělí a vytvářejí nové zdravé buňky. Jak keratinocyty stárnou a odumírají, tvrdnou a migrují do stratum corneum, kde vytvářejí ochrannou bariéru. Nakonec vypadnou, když je nahradí nově ztvrdlé buňky.
Při lamelární ichtyóze se kožní buňky pacienta vyvíjejí normálně, ale jak tvrdnou a migrují do stratum corneum, neodlupují se a brání jejich vypadávání. Nakonec se odumřelé kožní buňky hromadí a tvoří tvrdé, šupinaté pláty, které pokrývají tělo a charakterizují lamelární ichtyózu.
Ta nemoc je vzácná. Typicky se u pacientů rozvinou příznaky v raném dětství nebo se mohou narodit s šupinami. Příznaky mohou vymizet po většinu dospělosti a znovu se objevit později v životě.
Dermální porfyrie
Kutánní porfyrie ve skutečnosti tvoří šest různých typů dědičného onemocnění porfyrie. V každém z nich nejsou pacienti schopni produkovat enzymy, které vytvářejí hem, složku červených krvinek přenášející kyslík.
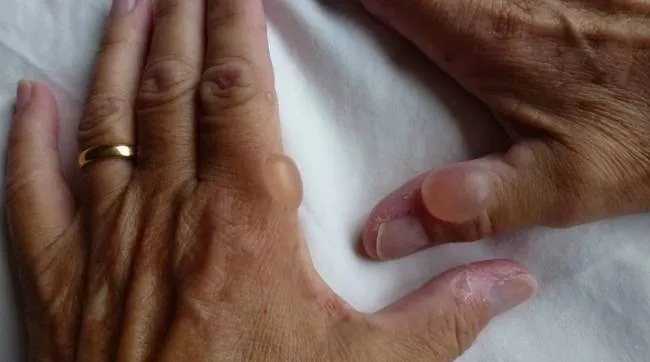
Hem se skládá z chemikálií zvaných porfyriny a ty se mohou hromadit, pokud nejsou přeměněny na hem. Nedostatek hemu a hromadění porfyrinů vede k příznakům porfyrie. Kožní porfyrie postihují kůži.
V případech kožní porfyrie je kůže pacienta extrémně citlivá na světlo. Po velmi krátkém pobytu na slunci se na kůži objeví zarudnutí, bolestivé podráždění a puchýře. Kůže může také nabobtnat při vystavení slunečnímu záření a abnormálně ztmavnout. V důsledku toho se pacientům důrazně doporučuje vyhýbat se slunečnímu záření.
Děti tuto nemoc zdědí od jednoho nebo více rodičů. Existuje osm různých enzymů, které přeměňují porfyriny na hem, a zděděná mutace v kterémkoli z genů, které exprimují tyto proteiny, může vést k porfyrii.
Keratoderma Maleda
Co se týče genodermatóz, de Maledova choroba patří mezi vzácnější typy. Toto dědičné kožní onemocnění se vyskytuje především u lidí středomořského původu a bylo pojmenováno podle ostrova Maleda, který se nachází nedaleko Chorvatska, kde byly zdokumentovány první případy.
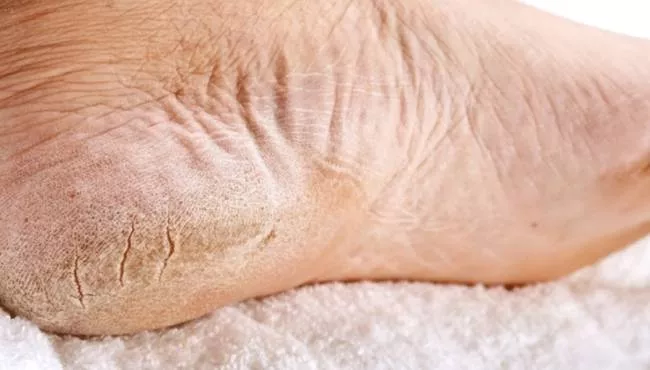
Toto onemocnění je druh keratózy dlaní a plosek kostí, častější typ kožního onemocnění charakterizovaný ztluštěním kůže na dlaních a ploskách nohou. Tento nárůst velikosti je výsledkem nárůstu velikosti kožních buněk. To zase způsobí, že se dlaně a chodidla zvětší a zežloutnou.
Protože de Maledova choroba je vzácná genodermatóza, není divu, že jde také o autozomálně recesivní onemocnění. To znamená, že musí být vloženy dvě kopie mutovaného genu, jedna od každého rodiče.
Gen SLURP1, který kóduje proteiny tvořící vazby mezi buňkami, byl identifikován jako viník de Maledovy choroby.
Pozornost! Informace uvedené v článku mají pouze informativní charakter. Materiály článku nevyžadují samoléčbu. Pouze kvalifikovaný lékař může provést diagnózu a poskytnout doporučení pro léčbu na základě individuálních charakteristik konkrétního pacienta.